
無菌製造とは何か
無菌製造とは、微生物や微粒子、その他の汚染物質がまったく存在しない環境で、注射薬のような直接体内に投与される医薬品を製造するプロセスです。経口薬とは異なり、注射薬は皮膚や消化管のバリアを通り抜け、直接血液中に投与されます。そのため、たった1つの細菌やウイルスが混入しただけで、敗血症や内毒素ショック、死亡に至る可能性があります。1920年代のインスリン汚染事件や1955年のカッター研究所のポリオワクチン事故、2012年のニューヨーク合併センターの髄膜炎アウトブレイク(751人感染、64人死亡)といった悲劇が、この分野の厳格な規制を生み出しました。
世界の規制基準
現在、無菌製造は複数の国際基準によって管理されています。アメリカ食品医薬品局(FDA)の21 CFR Part 210・211、欧州連合(EU)のGMP Annex 1(2022年改訂版)、世界保健機関(WHO)のTechnical Report Series No. 961、そしてISO 14644の清浄室基準が中心です。特に重要なのは、WHOが定める「汚染確率10^-6(100万個に1個以下)」というSterility Assurance Level(SAL)です。これは、100万個の製品のうち、たった1個でも微生物が混入してはならないという厳格な目標です。
清浄室の等級と環境管理
無菌製造は、清浄室(クリーンルーム)の等級によって段階的に制御されます。着衣室はISO 8(10万級)、作業エリアはISO 5(100級)が必須です。ISO 5エリアでは、0.5μm以上の微粒子が1立方メートルあたり3,520個以下でなければなりません。空気交換率は1時間に20〜60回、隣接する部屋との圧力差は10〜15パスカル、温度は20〜24℃、湿度は45〜55%に保たれます。水は「注射用水(WFI)」を使用し、エンドトキシンは0.25 EU/mL以下でなければなりません。容器は250℃で30分以上加熱して熱原を除去(デピロゲネーション)しなければなりません。
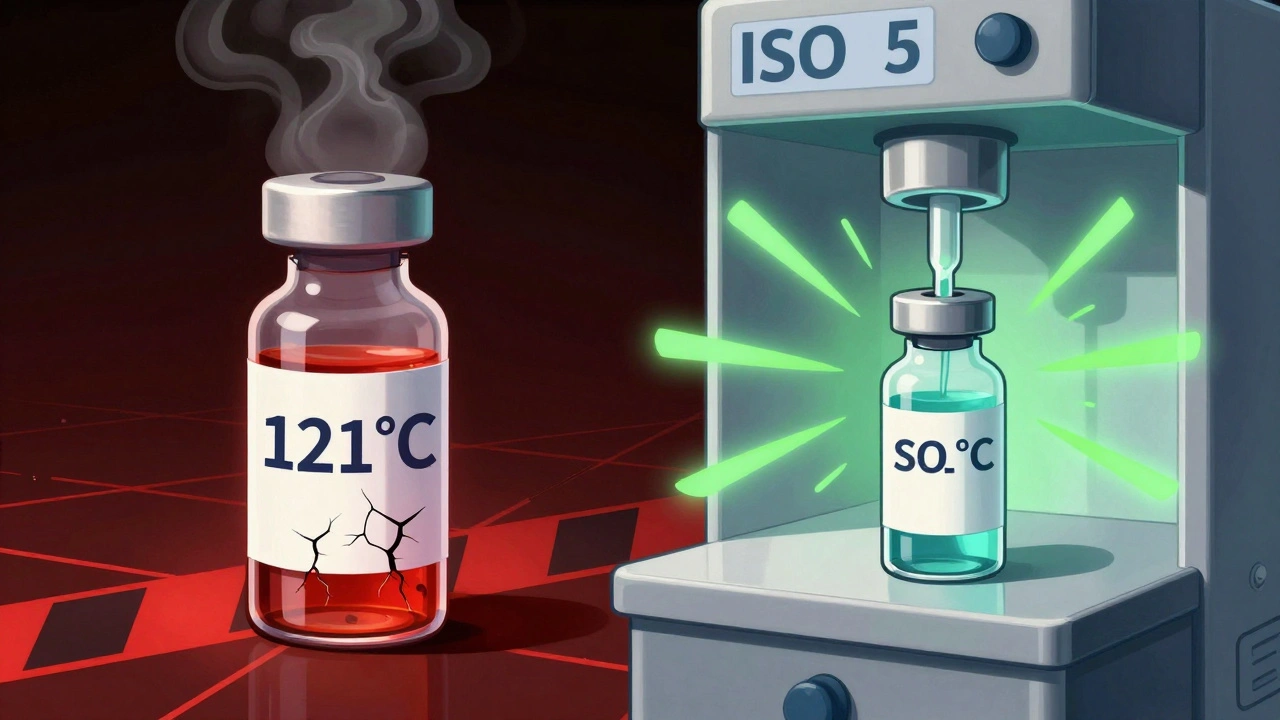
二つの製造方法:終末滅菌と無菌充填
注射薬の無菌製造には、主に二つの方法があります。
- 終末滅菌:製品を封じた状態で、121℃の蒸気で15〜20分間加熱したり、γ線で25〜50kGy照射して殺菌します。この方法はSAL 10^-6以上を達成でき、FDAが推奨します。しかし、熱や放射線に弱い生体薬品(モノクローナル抗体など)には適用できません。現在、注射薬の30〜40%しかこの方法で製造できません。
- 無菌充填:製品そのものを滅菌せず、製造全程で無菌状態を維持します。RABS(制限アクセスバリアシステム)やアイソレータと呼ばれる密閉型装置を使い、ISO 5環境を維持します。この方法は、生体薬品の90%以上に必要不可欠です。しかし、人為的ミスや環境汚染のリスクが高く、連続的な環境モニタリングが必須です。
環境モニタリングと検査
無菌充填では、空気中の微生物をリアルタイムで監視します。ISO 5エリアでは、空気サンプリングで1m³あたり1 CFU(コロニー形成単位)以下が「警戒レベル」、5 CFU以下が「対応レベル」とされています。1,000リットルの空気中に1 CFU以下という基準が、実際に製造中に達成されなければなりません。さらに、メディアフィル(培地充填)試験は、1回の検証で5,000〜10,000個の製品を充填し、そのうち0.1%以上で汚染が見つかったら、プロセスは不適切と判断されます。
コストと設備投資
終末滅菌の1バッチあたりのコストは約5万ドルですが、無菌充填は12万〜15万ドルと2〜3倍かかります。その理由は、高価なアイソレータやRABS、連続モニタリングシステム、そして厳格な人員教育にあります。小規模な無菌製造施設を新設するには、最低5,000万〜1億ドルの投資が必要です。人員は、年2回以上のメディアフィル試験に合格するまで、40〜80時間の無菌操作訓練を受けなければなりません。
現場の課題と失敗事例
業界の実態は厳しいです。FDAの2022年検査データによると、無菌製造の不適正の68%は「無菌操作の失敗」が原因です。一方、終末滅菌の問題は12%にすぎません。PDA(医薬品開発協会)の調査では、45の施設のうち68%が年1回以上、無菌検査で陽性反応を示していました。1回の失敗で平均120万ドルの損失が出ます。ある大手製薬会社の担当者は、RABSの手袋に亀裂が入り、3回のメディアフィルに失敗。45万ドルのロットが廃棄されたと報告しています。
最新の技術とトレンド
2023年以降、業界は大きく変化しています。閉鎖型プロセス(closed processing)の導入率は65%に上昇し、手作業による汚染リスクを大幅に削減しています。自動視覚検査装置の導入で、製品の欠陥率が0.2%から0.05%に低下した事例もあります。また、微生物検査の時間も、従来の14日間から24時間以内に短縮する「迅速微生物検査法」が広まり始めています。FDAは2024〜2026年の戦略で、AIを使ったデータ分析と自動監視を導入し、無菌製造の不適正を25%削減することを目標に掲げています。
市場の成長と今後の課題
2023年の世界の無菌注射薬市場は2,250億ドルに達し、年間8.2%の成長率で拡大しています。そのうち65%は生体薬品(特にモノクローナル抗体)が牽引しています。新薬承認の40%以上が無菌注射薬です。しかし、中国やインドなどの新興国では、FDAの検査に合格する施設がごくわずかです。2022年には、中国の1,200施設中、たった28施設しか合格していません。EUのGMP Annex 1(2022)では、従来の定期的なモニタリングから「連続モニタリング」への移行が義務化され、多くの製薬企業が1,500万〜2,500万ドルのアップグレード費用を負担しています。
成功の鍵は「継続的な管理」
無菌製造は、一度導入すれば終わりではありません。毎日、毎時間、毎分の管理が求められます。環境モニタリングデータ、人員のトレーニング記録、容器の洗浄・滅菌記録、そしてメディアフィルの結果--これらすべてが、患者の命を守る証拠になります。成功する企業は、単に規制を満たすのではなく、汚染を「防ぐ」文化を根付かせています。たとえば、ロンザのスイス工場では、連続モニタリングシステムを導入した結果、偏差が45%減り、製品のリリース速度が30%向上しました。これは、テクノロジーではなく、プロセスへのこだわりの勝利です。
無菌製造と普通の製造の違いは何ですか?
普通の製造(例:錠剤やカプセル)は、胃腸のバリアで微生物が除去されるため、比較的緩やかな清浄度で製造できます。しかし、注射薬は血液に直接入り込むため、微生物が1個でも混入してはいけません。そのため、清浄室の等級、環境モニタリング、人員訓練、プロセス検証のすべてが、通常の製造の10倍以上厳しくなります。
なぜ終末滅菌が使えない薬があるのですか?
生体薬品(モノクローナル抗体、遺伝子治療薬、ワクチンなど)は、熱や放射線に非常に弱いです。121℃の蒸気で加熱すると、タンパク質が変性して効果が失われます。そのため、これらの薬は、製造全程で無菌状態を保つ「無菌充填」方式でしか作れません。
メディアフィル試験とは何ですか?
メディアフィル試験は、無菌充填プロセスが本当に無菌であるかを検証するための実地テストです。実際の薬液の代わりに、微生物が育つ培地を充填し、その後、培養して汚染が起きなかったかを確認します。1回の試験で5,000〜10,000個の容器を充填し、0.1%以上で汚染が見つかると、そのプロセスは「不適切」と判断されます。
アイソレータとRABSの違いは何ですか?
アイソレータは完全密閉の箱型装置で、手袋を介して操作します。RABSは開放型のバリアで、操作者が手を出して作業できます。アイソレータは汚染リスクが低く、コストは高いです。RABSはコストが低く、操作性は良いですが、人為的汚染のリスクがやや高くなります。どちらも、正しく運用すれば同等の安全性を実現できます。
無菌製造の失敗は、なぜそんなに高価なのですか?
1バッチの損失は、単なる薬の廃棄ではありません。製造ラインの停止、再検証、再訓練、監査対応、規制当局からの是正要求、そして何より、患者への影響の可能性があります。平均1回の失敗で120万ドルの損失が出ますが、ブランド信頼の損失や訴訟リスクは、金額では計れません。




コメント
門間 優太
13 12月 2025この記事、めっちゃわかりやすかった。無菌製造の厳しさを改めて実感した。人間の手が入る部分が一番リスク高いってところ、本当にそうだと思う。
Hideki Kamiya
14 12月 2025おいおい、FDAが言ってるってことは…ってことは政府が隠してるんだよな?10^-6って、あんまりにも完璧すぎて逆に疑わしい。本当は全部監視されてて、汚染されてるのに隠してるんだろ?💀
Keiko Suzuki
15 12月 2025無菌製造の重要性について、非常に丁寧にまとめられていて感謝します。特にメディアフィル試験の基準や、RABSとアイソレータの違いの説明は、現場で働く方々にとっても参考になると思います。
花田 一樹
15 12月 2025120万ドルのロットが廃棄って、結局は誰が払ってるの?患者?保険?それとも税金?🤔
EFFENDI MOHD YUSNI
16 12月 2025この文脈における「汚染確率10^-6」は、単なる統計的幻想である。現実の清浄室では、人間の呼吸、皮膚剥離、衣類の繊維が常に動的汚染源となり、理論値と実績の乖離は不可避。これはシステムの欺瞞である。
JP Robarts School
17 12月 2025中国の1200施設で28施設しか合格してないって…ってことは、世界の薬の半分以上は危険ってこと?誰かが死ぬまで気づかないんだろうな。
Mariko Yoshimoto
18 12月 2025いや、でも…、ISO 5の微粒子数って、3,520個/㎥って、本当に正確?だって、人間の皮膚から毎分100万個以上剥がれてるって、文献で読んだことあるし…?????
HIROMI MIZUNO
19 12月 2025AI監視導入で25%削減って、めっちゃ希望持てる!これで現場の人が少しでも楽になればいいな😊
晶 洪
20 12月 2025人間が関わる限り、無菌は無理。
naotaka ikeda
20 12月 2025終末滅菌が使えない薬が増えている現実。生体薬品の需要が高まるほど、無菌充填のリスクも増える。このバランスをどう取るかが、次の10年の課題。
諒 石橋
22 12月 2025海外の製薬会社が日本より安いって言ってるけど、それって日本が過剰に安全にしすぎてるだけだろ?中国の製薬工場で作っても、ちゃんと検査すればいいだけじゃん。
risa austin
23 12月 2025本件に関する規制の進化は、医療倫理の根本的な再定義を伴うべきである。すなわち、生命の尊厳に対する社会的合意が、技術的基準を上回る必要性が顕在化している。
Taisho Koganezawa
23 12月 2025もしAIが汚染をリアルタイムで予測できるなら、人間の判断は不要になる。でも、それって結局、人間が作ったシステムが人間のミスを補うって話で、根本的な矛盾だよね?
Midori Kokoa
25 12月 2025メディアフィルで0.1%汚染って、もうちょっと緩くてもいいのでは?