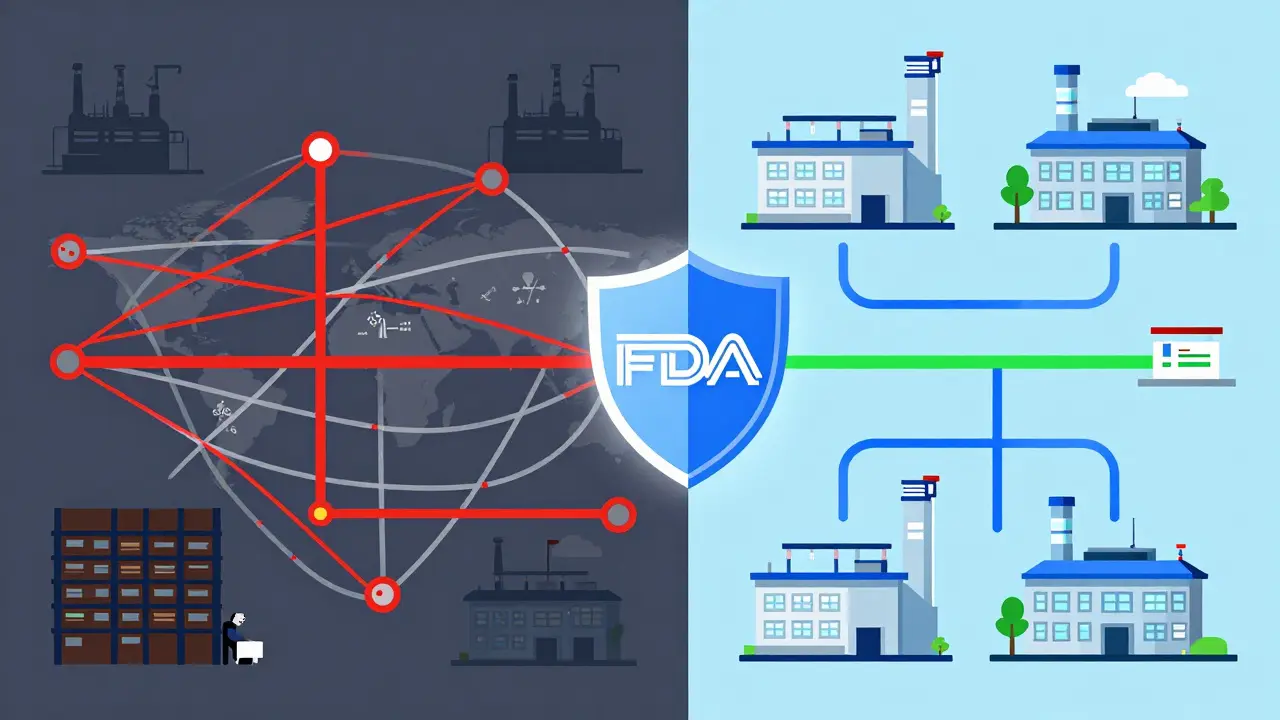
アメリカ食品医薬品局(FDA)のジェネリック医薬品承認プロセスは、2023年から2025年にかけて劇的な変化を迎えています。かつては「コスト削減」と「迅速な市場投入」が最優先されたこの分野ですが、今では「国家安全保障」と「供給チェーンの強靭化」が最上位の課題に浮上しました。特に2025年10月に発表されたANDA Prioritization Pilot Program(略称:ANDA優先パイロットプログラム)は、2012年のGDUFA以来、最も包括的な制度改革と言えます。
この変更は何を意味するのでしょうか? 単なる官僚主義の変更ではありません。これは、米国が外国依存から脱却し、国内での医薬品製造を再興するための戦略的転換点です。もしあなたが製薬企業の関係者、投資家、あるいは医療政策に関心のある方であれば、これらの動きがあなたのビジネスや業界全体に与える影響を理解することは必須です。
ANDA優先パイロットプログラムの概要と背景
FDAが2025年10月3日に発表したANDA Prioritization Pilot Programは、米国内で試験および製造を行うジェネリックメーカーに対して、審査期間を大幅に短縮するというインセンティブを提供します。このプログラムの根底にあるのは、パンデミック中に露呈した供給チェーンの脆弱性への対応です。
現在の状況を見ると、驚くべきデータがあります。2025年時点で、活性医薬成分(API)メーカーのうち米国内に所在するものはわずか9%です。対照的に、中国は22%、インドは44%を占めています。さらに、米国で流通する医薬品の半数以上が海外で製造されています。このような状況下で、外国施設の検査には国内検査よりも47%多くの準備時間が必要で、コストも32%高くなるとFDAのジョージ・ティドマーシュ博士は指摘しています。
このプログラムは、MAPP 5240.3という手続規則に基づいて運営され、国内製造とテストの検証に基づいてANDA(略式新薬申請)の審査、修正、補充申請の優先順位を決定します。これは、トランプ政権の「アメリカンファースト・ファーマ」イニシアチブの一環として、政治的にも強力な支持を得ています。
審査プロセスの変化:速度と効率の追求
従来のANDA審査と比較すると、パイロットプログラム参加者のメリットは明確です。標準的な審査サイクルが12〜15ヶ月であるのに対し、Tier 1(100%国内製造・テスト)の対象となる申請は、目標8ヶ月の審査サイクルを目指しています。
| 指標 | ANDA優先パイロット (Tier 1) | 従来型ANDA審査 |
|---|---|---|
| 初期審査開始まで | 30日以内 | 60〜90日 |
| 完全回答書(CRL)発行 | 提出後45日以内 | 提出後120日以内 |
| 平均市場投入までの時間 | 11.2ヶ月 | 15.6ヶ月 |
| 初回承認率 | 33%高い | 基準値 |
| 主要欠陥通知の発生率 | 41%減少 | 基準値 |
FDAの推計によると、このプログラムにより適格な申請の審査時間は35〜40%短縮されます。また、国内製造能力は3年間で22%増加すると見込まれています。ただし、複雑なジェネリック(狭治療域指数薬や特定の経皮吸収パッチなど)は当初のパイロット段階からは除外されており、2026年1月の拡大を待つ必要があります。

経済的影響とコスト構造の変化
速度が向上する一方で、コスト面での課題も無視できません。国内製造施設を設立するための資本投資は、中規模のジェネリック生産施設で1億2,000万〜1億8,000万ドルと見積もられています。さらに、国内施設検証のための追加コストとして、申請件あたり120万〜180万ドルがかかります。
ジェネリック医薬品協会(GPhA)の報告によれば、完全に国内サプライチェーンを持つメーカーのパイロット下での承認成功率は92%ですが、外国製造要素を含む申請は68%にとどまります。しかし、高用量・低利益のジェネリックでは、外国製造が依然として25〜30%のコスト優位性を保っています。
MedPAC(メディケア支払諮問委員会)の2025年6月報告書は、国内製造要件によりジェネリック医薬品の価格が当初12〜18%上昇する可能性があると警告していますが、国内容量が拡大する3〜5年後には正常化するとの予測を示しています。長期的には、医薬品不足や緊急調達コストの削減により、2030年までに年間42億ドルの純節約が見込まれ、納税者にとって2027年には費用対効果中立になると国会予算办公室(CBO)は分析しています。
業界の声:賛否両論と実態
業界内の反応は一様ではありません。テバ・ファーマシューティカルズなどの大手企業は、nimodipine溶液などを計画より8ヶ月早く市場に投入できたことを評価しています。一方、中小企業からは資金圧迫への懸念が強まっています。
r/pharmaceuticalsでの2025年8月の議論では、製造業者の68%が長期的な利益に楽観視しているものの、79%が即座のコスト圧力、特に限られた資本を持つ小規模企業における問題を指摘しました。アクセス可能医薬品協会(AAM)の調査でも、回答者の54%が国内施設の拡張を開始した一方、31%が移行コストのために特定製品の開発を延期していると報告しています。
ハーバード医科大学のアロン・ケッセルハイム教授は、JAMA Internal Medicine(2025年3月)で、パイロットプログラムを通じた加速されたジェネリック承認が、伝統的に承認されたジェネリックと比較して同等の治療結果(主効能エンドポイントに対する95%信頼区間:0.97-1.03)を維持していることを示す研究を発表しました。これにより、安全性に関する懸念は科学的に裏付けられています。

今後の展望と複雑なジェネリックへの拡大
2026年1月から、パイロットプログラムは複雑なジェネリックにも拡大されます。鼻スプレー、眼科用懸濁液、経皮吸収パッチなどへの具体的なガイダンスは、2025年11月に予定されています。また、AI支援レビュープロトコルの導入により、パイロット申請の審査時間がさらに25%短縮される可能性があります。
FDAのジェネリック医薬品事務所は、2028年までに国内API製造シェアを9%から23%に引き上げると予測しています。しかし、欧州ジェネリック医薬品協会が2025年7月に公式照会を出したように、WTOの貿易適合性に関する潜在的なリスクも存在します。これらの国際的な摩擦は、今後数年間の医薬品貿易において重要な争点になるでしょう。
実践的なアドバイス:メーカーと関係者へ
これらの変化に対応するため、メーカーは国内サプライチェーン管理と規制戦略における新しい能力を開発する必要があります。外国から国内製造モデルへの移行には、平均4〜6ヶ月の学習曲線が必要です。FDAによると、初期のパイロット申請の82%が、不十分な国内検証書類のため再提出を余儀なくされました。
必要なスキルには、FDAの製品別ガイドライン(PSG)の詳細な知識が含まれます。2024-2025年には、パイロット要件に合わせて78のPSGが更新されました。一般的な課題としては、API品質の一貫性(欠陥通知の67%)、複雑なジェネリックのバイオベクвивалент研究デザイン(43%)、安定性試験プロトコル(38%)が挙げられます。FDAはこれらの問題を解決するために専任の技術支援チームを設置し、30日以内に89%の問題を解決しています。
ANDAとは何ですか?
ANDA(Abbreviated New Drug Application)は、新薬申請(NDA)ほど詳細な臨床試験データを必要とせず、既存の新薬と同じ有効性・安全性であることを証明することで、ジェネリック医薬品の承認を得るための申請手続きです。これが今回の改革の中心となっています。
なぜFDAは国内製造を優先するのですか?
パンデミック中に顕在化した供給チェーンの脆弱性と、外国依存による国家安全保障上のリスクを軽減するためです。また、外国施設の検査にかかる時間とコストの高さも理由の一つです。
ANDA優先パイロットプログラムに参加するにはどうすればよいですか?
米国内でCGMPに準拠した施設での製造と、FDA登録ラボでの国内バイオベクвивалентテストを実施し、その証拠を提出する必要があります。複雑なジェネリックは2026年以降の対象となります。
この変更は薬価に影響しますか?
短期的には、国内製造のコスト増により12〜18%の価格上昇が見込まれます。しかし、長期的には供給安定化による緊急調達コスト削減などで、社会全体のコスト削減効果が期待されています。
複雑なジェネリックはいつ対象になりますか?
2026年1月からプログラムが拡大され、鼻スプレーや経皮吸収パッチなどの複雑なジェネリックも対象に入ります。具体的なガイドラインは2025年11月に発表予定です。





コメントを書く